(Editors’ Note: This article covers a stock trading at less than $1 per share and/or with less than a $100 million market cap. Please be aware of the risks associated with these stocks.)
We have decided to take a very detailed look at iCo Therapeutics (ICO.V) (OTCQX:ICOTF) and the company’s Phase 2 drug for Diabetic Macular Edema (DME), iCo-007, an anti-sense drug candidate in-licensed from ISIS Pharmaceuticals in 2005. Our research is presented below. Our sum-of-parts analysis tells us that iCo’s stock has 125% upside based on a 50% probability of success of iDEAL in April 2014. Comparator analysis tell us that the shares could be worth as much as $3.00, or +633% upside, if iDEAL hits on the full data analysis in September / October 2014. As such, we see meaningful upside in owning the stock at this level.
What Is Diabetic Macular Edema?
According to the American Diabetes Association, 25.8 million Americans have diabetes. A recent study out of the Singapore Eye Research Institute suggests that 7% of diabetic patients may develop diabetic macular edema (DME) or diabetic retinopathy (DR) (Ding J. et al, 2012). The risk factors for DME are largely similar to DR, but dyslipidemia appears to play a more significant role. Applying the 7% prevalence suggested by Ding J. et al pegs the U.S. DME market at around 1.8 million people. Another study out of the Lawson Health Research Institute in Canada found an approximate 15.7% prevalence of DME among diabetic patients, with 2.56% having significant visual impairment (Petrella RJ. et al, 2012). This suggest a U.S. population of nearly 4 million patients, 0.65 million with severe visual impairment.
DME occurs when blood vessels in the retina of patients with diabetes begin to leak into the macula, the part of the eye responsible for detailed central vision. These leaks cause the macula to thicken and swell, progressively distorting acute vision. While the swelling may not lead to blindness, the effect can cause a severe loss in central vision. Nevertheless, DME is the major cause of vision loss in people with diabetic retinopathy.
The retina is the light sensitive area at the back of the eye, and the macula is the central part of the retina used for high resolution vision. In diabetes, the accumulation of metabolic products arising from the processing of excess serum glucose leads to capillary damage, which has multiple sight-threatening consequences. The leakage of damaged capillaries causes proteins and extracellular fluids to build up in the retina, leading to swelling (edema). Edema within the macula causes a blurring of vision.
Hypoxia caused by capillary damage causes the release of vascular endothelial growth factor (VEGF) and other pro-angiogenic factors. The new blood vessels formed in response are poorly formed and fragile, accompanied by fibrous supporting tissue, and may invade the vitreous fluid. Bleeding from these fragile blood vessels may create blind spots by blocking light from reaching the retina, and the fibrous support stresses the eye structure and may lead to retinal detachment and a profound loss of vision. VEGF also promotes vascular permeability, thus exacerbating macular edema.
Diabetic retinopathy becomes nearly ubiquitous with long-standing diabetes. After 20 years with the disease, 60% of Type-2 diabetics and virtually 100% of Type-1 diabetics will manifest some form of retinopathy. Alpharetta, Georgia-based Alimera Sciences (NASDAQ:ALIM), estimated diabetics have a 10% risk of developing the condition during their lifetime; this is consistent with the data from Singapore and Canada noted above. Given the large and rapidly growing U.S. pre-diabetic population, we estimate that approximately 300,000 new cases of DME develop annually. California-based Genentech, estimates that 55% of Americans with DME are unaware they have the disease, and that 70% of the patients with diabetic retinopathy will go on to develop DME.
…Current Treatment Options…
Laser photocoagulation: The mainstay of therapy for diabetic macular edema is laser photocoagulation. Compromised spots within the blood vessels of the retina are specifically targeted with the laser, which causes clots and halts fluid leakage. Ultimately, this leads to improved oxygen supply to the retina and reduced downstream neovascularization. Microaneurysms, the sources of leakage in DME, are targeted by the laser, and hemoglobin in the microaneurysms absorbs the laser energy. This promotes thrombosis within the microaneurysm, halting further leakage. Clinical trials have demonstrated a 50% reduction in moderate to severe visual loss from DME when laser photocoagulation is initiated early in the course of the disease.
A 10-year study by the National Eye Institute called Early Treatment Diabetic Retinopathy Study (ETDRS) was designed to evaluate the effectiveness of both argon laser photocoagulation and aspirin therapy in delaying or preventing progression of early diabetic retinopathy to more severe stages of visual loss and blindness. The study was also designed to help determine the best time to initiate photocoagulation treatment, to monitor closely the effects of diabetes mellitus and of photocoagulation on visual function, and to produce natural history data that can be used to identify risk factors and test etiologic hypotheses in diabetic retinopathy. A total of 3,711 patients were followed for a minimum of 4 years to provide long-term information on the risks and benefits of the treatments under study. Results of ETDRS suggest (source: NEI/NIH, 1999):
- Aspirin use did not affect the progression of retinopathy to the high-risk proliferative stage in eyes.
- Photocoagulation reduced the risk of moderate vision loss, especially for those eyes with macular edema that involved or threatened the center of the macula. There was moderate visual gain in those eyes that received focal treatment as well as a decrease in the amount of retinal thickening.
- A statistically significant reduction in severe visual loss was found for those patients with early treatment, especially for those patients with non-insulin-dependent diabetes mellitus.
The advantages of photocoagulation have been made clear by the ETDRS. However, macular photocoagulation is not without risks. Complications of macular laser treatment include paracentral scotomas, lateral creep of juxtafoveal laser scars into the fovea, accidental foveal photocoagulation, subfoveal fibrosis, and choroidal neovascularization at the sites of laser scars. In addition, there can be residual massive hard exudates after the resolution of edema, and patients often experience color vision impairment.
In general, laser photocoagulation prevents further vision loss but does not routinely restore vision already lost to DME. As such, photocoagulation is commonly supplemented by medical treatments, among which the intravitreous administration of corticosteroids and vascular endothelial growth factor antagonists are the most widely used.
Corticosteroids: Use of corticosteroids is common in the treatment of macular edema. There are a handful of corticosteroid-based intravitreal implants under development. Products include a dexamethasone biodegradable implant (Posurdex®, Allergan), the helical triamcinolone acetonide implant (I-vation™ TA, SurModics), the fluocinolone acetonide implant (Retisert®, Bausch Lomb), and another fluocinolone acetonide-based implant that is injectable (Medidur™, pSivida / Alimera Sciences). Although not a novel concept, drug delivery via intravitreal implant remains undesirable to patients. The advent of sustained release corticosteroids has made repeated intravitreal injections unnecessary, and thus use of corticosteroids has grown over the last decade.
Iluvien™ (fluocinolone acetonide) is a drug-device combination product approved in the UK, Austria, France, Germany, Portugal, and Spain for the treatment of DME. Iluvien™ is an injectable, non-erodible, intravitreal implant for the treatment of DME that works by slowly releasing the corticosteroid drug fluocinolone acetonide for up to three years after implant. The device is designed to be small enough to be injected into the back of the eye with a 25 gauge needle creating a self-sealing hole. This insertion procedure is very similar to an intravitreal injection for other AMD or DME products. Iluvien™ has been rejected by the U.S. FDA on three separate occasions, the third and most recent of which took place in October 2013 and seemingly killed the drug.
However, in December 2013, the U.S. FDA threw a life-line to Alimera and partner pSivida (PSDV) by entering into an agreement to cancel a planned January 2014 Dermatologic and Ophthalmic Advisory Committee meeting in return for labeling discussions on the drug. As of the last update, Alimera will focus instead on drafting its response to the Complete Response Letter (CRL) received from the FDA in October 2013, with a goal of submitting the response in the first quarter of 2014.
According to Alimera, the response intends to address concerns the FDA raised regarding the facility at which Iluvien™ is manufactured. In addition, Alimera expects to provide a safety update and additional data on the product from patients and from physician experience with the applicator in the U.K. and Germany, where Iluvien is currently commercially available. The FDA has indicated that Alimera will not be required to conduct any new clinical trials in connection with the FDA’s review prior to approval.
Clinical data on Iluvien™ was recently published in the February 2013 issue of Drug (Vol. 73, Issue 2, 187-93). Results from two multinational trials in patients with DME previously treated with macular laser photocoagulation, fluocinolone acetonide intravitreal implant 0.2 μg/day was significantly more efficacious than sham injection in improving visual acuity. At 24 months post injection, 29% of fluocinolone acetonide intravitreal implant 0.2 μg/day recipients had an improvement in the best-corrected visual acuity (BCVA) letter score of ≥15 compared with 16% in the sham injection group (p=0.002).
Treatment benefit was most evident in the subgroup of patients whose duration of DME was ≥3 years. In this subgroup at 36 months, 34% of fluocinolone acetonide intravitreal implant 0.2 μg/day recipients had an increase in the BCVA score of ≥15, compared with 13% of sham injection recipients (p0.001).
Iluvien™ looks like a far less effective drug than Eylea® (discussed below). Iluvien™ achieved a “percent of patients achieving a BCVA ≥15 letter score” of 29% at the end of two years compared to Eylea that seems to achieve 56-60% at the end of only 24 weeks. The benefits of Iluvien™ are the one-time injection that last up to three years. For a three-year treatment with Eylea®, a DME patient might expect 22-36 injections.
However, high incidence of intraocular pressure (IOP, 21 mmHg) may limit Iluvien™ uptake. For example, the products UK prescribing label notes the proportion of Iluvien™ treated subjects requiring treatment with IOP lowering medication was 38% compared to 14% in the sham treated group. This proportion increased to 47% in those subjects with greater than median IOP at baseline (≥15 mmHg). Surgical interventions for the treatment of ocular hypertension were required in 4.8% of subjects treated with Iluvien™ compared to 0.5% of subjects treated with sham. Patients with high baseline IOP must be closely monitored when using Iluvien™. Besides high IOP, results from the FAME study show incidence of cataract surgery among all patients was approximately 3-fold higher for the Iluvien® group (80.0%) than in the sham group (27.3%).
With Lucentis® and Eylea® doing an estimated $1.3 billion and $650 million worldwide in DME, with about half the sales coming from the U.S., Iluvien™ looks like a $200 million U.S. and $100 million Ex-U.S. drug. Nevertheless, the Iluvien™ data and tenuous path to market in the U.S. represents an interesting benchmark for the development of a new treatment for DME.
VEGF-Inhibitors: Use of VEGF inhibitors have gained significant steam over the past decade since the approval of Genentech’s Lucentis (ranibizumab) for age-related macular degeneration in June 2006, and the subsequent off-label use of Genentech’s oncology drug, Avastin® (bevacizumab), a far cheaper and similar efficacy alternative. The properties of VEGF, and the consequences of its inhibition, also suggest a role for this approach in the management of DME. The mechanism of action for Lucentis® and Avastin®, inhibition of VEGF and other insulin-like growth factors that mediate angiogenesis, protease production, endothelial cell proliferation, migration, and tube formation is of similar concept in both oncology and ophthalmology indications. VEGF increases vascular permeability by relaxing endothelial cell junctions, which increases permeability and leakage. Inhibition of VEGF blocks this effect to some extent, as demonstrated in several recent clinical trials and case series involving the anti-VEGF molecules pegaptanib, ranibizumab, and bevacizumab.
- Pegaptanib sodium (Macugen®, Valent Pharma) is an anti-VEGF aptamer, a small piece of RNA that self-folds into a shape that binds to and blocks the effects of VEGF165, one isoform of the VEGF family of molecules. The drug is approved by the FDA for the treatment of age-related macular degeneration. Results from a mid-stage DME study demonstrate that a modest mean improvement in baseline visual acuity in the pegaptanib 0.3-mg group versus a control. We regard the result as unimpressive.
- Ranibizumab (Lucentis®, Genentech) is an antibody fragment that also binds and blocks the effects of VEGF. Unlike pegaptanib, ranibizumab binds and inhibits all isoforms of VEGF. Lucentis® is approved by the FDA for the treatment of age-related macular degeneration and diabetic macular edema. We discuss the efficacy of ranibizumab below.
- Bevacizumab (Avastin®, Genentech) is the full antibody from which ranibizumab is derived. This anti-VEGF molecule is FDA approved for systemic treatment of metastatic colon cancer, but not for any ophthalmic indications. Its use in conditions such as age-related macular degeneration, diabetic retinopathy, and DME is currently off-label. The efficacy of Avastin® in AMD is comparable to that of Lucentis®. We discuss this below.
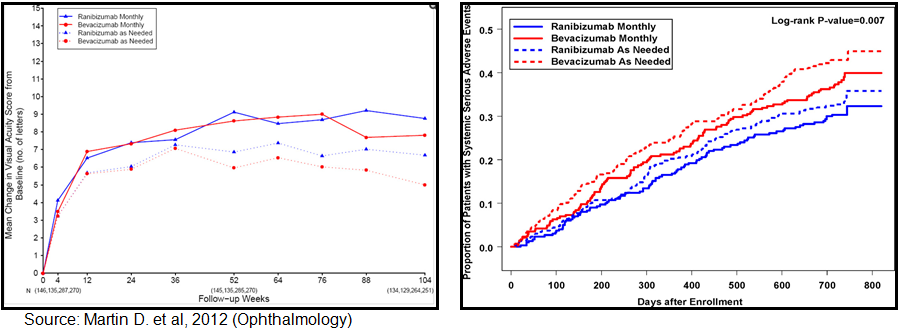
Avastin® vs. Lucentis®: Randomized clinical trials have been conducted comparing bevacizumab to ranibizumab for neovascular age-related macular degeneration. Results have been published in the New England Journal of Medicine (May 2011) and the Journal of Ophthalmology (July 2012). A total of 1,185 patients with neovascular age-related macular degeneration were enrolled in the clinical trial. A total of 1,107 of the 1,185 were followed for two years. At enrollment, patients were assigned to 4 treatment groups defined by drug (ranibizumab or bevacizumab) and dosing regimen (monthly or as needed). At 1 year, patients initially assigned to monthly treatment were reassigned randomly to monthly or as-needed treatment, without changing the drug assignment.
According to published results, among patients following the same regimen for 2 years, mean gain in visual acuity was similar for both drugs (bevacizumab-ranibizumab difference, -1.4 letters; 95% confidence interval [CI], -3.7 to 0.8; p=0.21). Mean gain was greater for monthly than for as-needed treatment (difference, -2.4 letters; 95% CI, -4.8 to -0.1; p=0.046). The proportion without fluid ranged from 13.9% in the bevacizumab-as-needed group to 45.5% in the ranibizumab monthly group (drug, p=0.0003; regimen, p0.0001). Switching from monthly to as-needed treatment resulted in greater mean decrease in vision during year 2 (-2.2 letters; p=0.03) and a lower proportion without fluid (-19%; p=0.0001). Rates of death and arteriothrombotic events were similar for both drugs (p=0.60). The proportion of patients with 1 or more systemic serious adverse events was higher with bevacizumab than ranibizumab (39.9% vs. 31.7%; adjusted risk ratio, 1.30; 95% CI, 1.07-1.57; p=0.009). Most of the excess events have not been associated previously with systemic therapy targeting vascular endothelial growth factor (VEGF).
{kind=link}
Ranibizumab and bevacizumab had similar effects on visual acuity over a 2-year period. Treatment as needed resulted in less gain in visual acuity, whether instituted at enrollment or after 1 year of monthly treatment. There were no differences between drugs in rates of death or arteriothrombotic events. The interpretation of the persistence of higher rates of serious adverse events with bevacizumab is uncertain because of the lack of specificity to conditions associated with inhibition of VEGF. The primary impetus for significant off-label use of bevacizumab is cost. According to Ron Afashari, MD of the Yale School of Medicine and Director of the Yale Retina Service, Avastin® (bevacizumab) costs around $30 to $50 per dose when used off-label, whereas Lucentis® (ranibizumab) costs around $2,000 per dose. Roche sold an estimated $4.0 billion worth of Lucentis® in 2012. We estimate that approximately 1/3rd of the Lucentis® sales are off-label in DME.
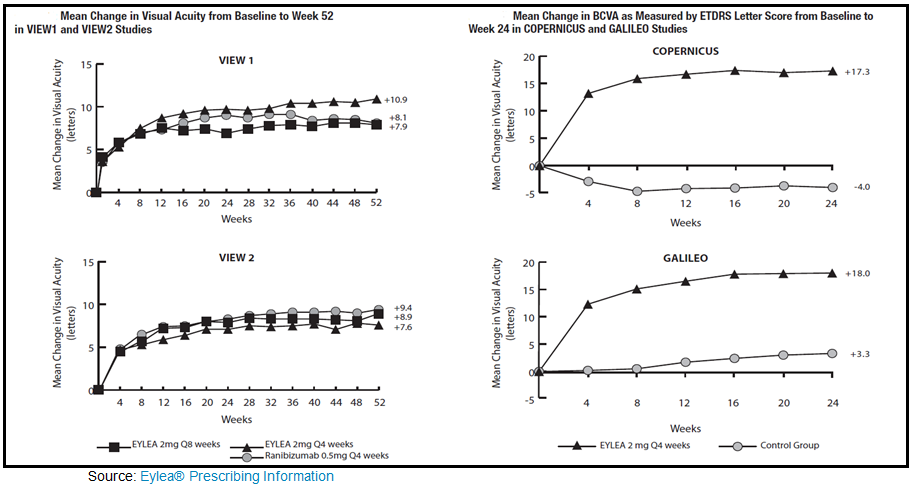
Aflibercept (Eylea®, Regeneron Pharmaceuticals) injection is a relatively new product for the treatment of (wet) age-related macular degeneration in November 2011. Since its approval, Eylea has gained significant market share on Lucentis® and Avastin® for AMD and off-label use in DME, primarily because the drug can be given every 4 weeks as opposed to the weekly injections with the aforementioned two. In fact, after the first 12 weeks of treatment, dosing can be titrated down to every 8 weeks. The drug is also approved for use in the treatment of patients with Macular Edema following Central Retinal Vein Occlusion (CRVO). The recommended dose for Eylea is 2 mg administered by intravitreal injection every 4 weeks (monthly) for five weeks, then every 8 weeks as a maintenance therapy. Eylea® 2013 worldwide sales are estimated at approximately $1.8 billion, with $1.4 billion in the U.S.
To gain approval, Regeneron conducted two Phase 3 studies in AMD, VIEW-1 and VIEW-2, and two additional Phase 3 studies, COPERNICUS and GALILEO in macular edema following CRVO. The data below highlights the primary efficacy findings for Eylea in both AMD and MD-CRVO as listed in the prescribing insert:
{kind=link}
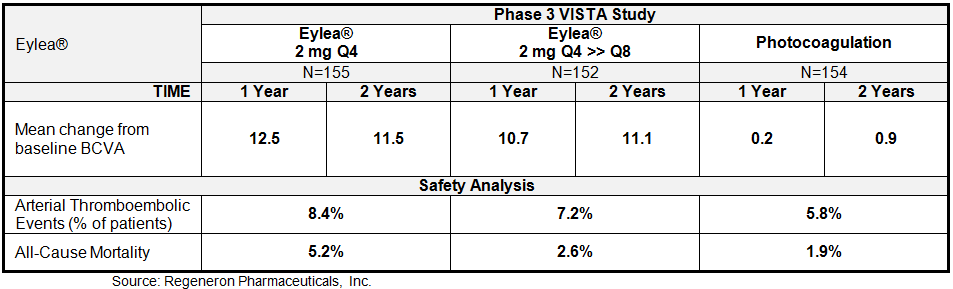
Regeneron just recently reported results from the 100-week follow-up analysis from the Phase 3 VISTA trial studying Eylea® for DME. In this trial, patients with DME were randomized to receive either Eylea® monthly (2Q4: n=155), Eylea® monthly for five months, then every two months as a maintenance (2Q8: n=152), or the comparator treatment of laser photocoagulation (n=154). Results showed a sustained improvement from baseline in best corrected visual acuity (BCVA) at week 100, compared to laser photocoagulation. The data at 52-weeks was previously reported, and included below.
{kind=link}
We see the results from the VISTA study as particularly encouraging given that 43% of patients in this study had previously received anti-VEGF therapy. Besides demonstrating impressive improvement in BCVA after 100 weeks of treatment, Eylea® was generally well tolerated with a similar overall incidence of adverse events (AEs), ocular serious AEs, and non-ocular serious AEs across similar between the three cohorts. AEs were typical of those seen in other studies in patients with diabetes receiving intravitreal anti-VEGF therapy. The most frequent ocular AEs observed in the VISTA-DME trial included conjunctival hemorrhage, eye pain, and vitreous floaters. The most frequent non-ocular AEs included hypertension, anemia, and urinary tract infection. Overall, the data for Eylea® compares well with Lucentis®. Adverse events for the two drugs are pretty similar, with conjunctival hemorrhage (25% vs. 28%), eye pain (9% for both), cataract (7% for both), and vitreous detachment and floaters (6-7% for both) the most common in the AMD studies. Eye pain (13% vs. 5%), conjunctival hemorrhage (12% vs. 11%), and intraocular pressure (8% vs. 6%) were the most common in the MD-CRVO studies.
Regeneron plans to present full two-year data from the VISTA-DME trial at upcoming medical conferences. Two-year data from the similarly designed VIVID-DME trial are expected later in 2014. Both the VISTA-DME and the VIVID-DME trials will continue as planned up to 148 weeks. Regeneron has filed regulatory submissions in the U.S. and the EU for Eylea® for the treatment of Diabetic Macular Edema.
…Best Available Care – Based On Data Cost…
In December 2013, a detailed paper was published in Therapeutic Advances in Endocrinology and Metabolism by Boyer D.S., et al (Vol. 4(6): 151-169) highlighting the data from all completed randomized Phase 2 or Phase 3 clinical trials with anti-VEGF therapy in patients with DME. The authors conclude:
The previous standard of care for patients with DME has focused on the prevention of further deterioration in vision using macular laser once some degree of loss has already occurred, with very few patients experiencing any subsequent gains in vision. Studies of the pathophysiology of DME demonstrate a crucial role for VEGF in disease development and this has led to successful clinical trials of VEGF inhibitors. Data from several large, prospective randomized clinical trials indicate that, on average, intraocular inhibition of VEGF is associated with rapid resolution of DME(as indicated by reduction of the thickness of the retina) and significant VA gains, better than those achieved with focal/grid laser photocoagulation, demonstrating that the focus of treatment should be on improvements in vision and not the prevention of further worsening.
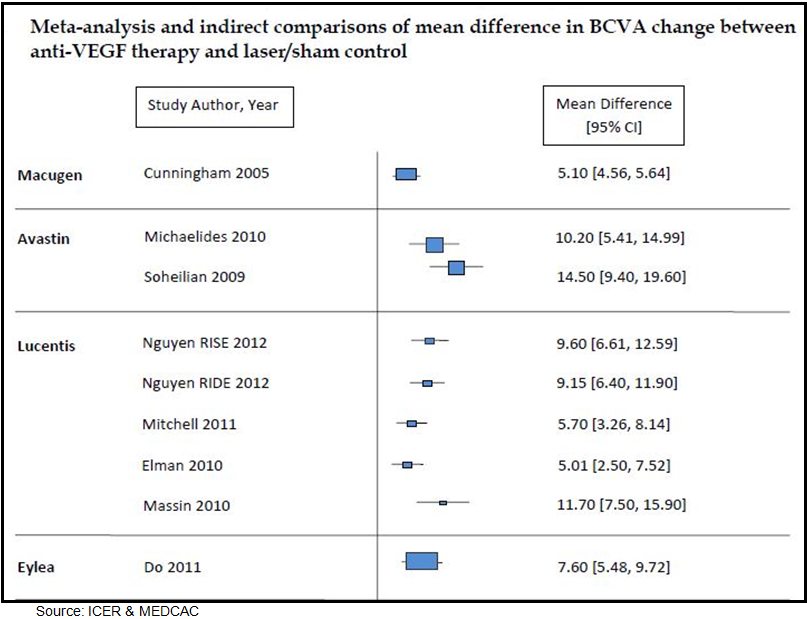
In May 2012, the Institute for Clinical and Economic Review (ICER) prepared a technology assessment report on anti-vascular endothelial growth factor treatment for diabetic macular edema. The report was prepared for the Medicare Evidence Development Coverage Advisory Committee (MEDCAC). ICER analyzed a total of 15 randomized clinical trials and eight observational studies. The authors of the report concluded that anti-VEGF therapy improves visual acuity in patients with DME relative to macular laser treatment of sham injection. However, no significant difference between the anti-VEGF agents was found in terms of clinical performance, and the authors cautioned that the unknown systemic side-effect profile of drugs like Avastin® relative to Lucentis® and Eylea® remains the greatest element of uncertainty.
Below is a figure from the report highlighting the prospective efficacy findings from “fair” or “good” quality studies with anti-VEGF therapy.
{kind=link}
Based on the results of the head-to-head analysis conducted by Martin D. et al, 2012 (Ophthalmology) and the technology assessment report prepared by ICER for MEDCAC, off-label use of Avastin® (bevacizumab) seems like the most effective anti-VEGF therapy for DME. Efficacy results look similar to Lucentis® (ranibizumab), with cost for yearly treatment favoring the off-label bevacizumab by nearly 40-to-1. We estimate one year treatment of Lucentis® (at approximately $2,000 per dose) costs $18,000 to $24,000, whereas yearly treatment with off-label use of bevacizumab (currently priced at around $50 per dose) costs only $450 – $600.
Regardless of the efficacy and economics that seem to favor off-label use of Avastin®, Lucentis® and Eylea® are generating over $3.3 billion in sales in DME. The systemic side-effect profile of Avastin® and the lack of physician desire to dilute out off-label bevacizumab, as well as aggressive marketing by Roche, is what are driving use of Lucentis®. We believe use of Eylea®, priced at roughly a 5% discount to Lucentis®, is being driven by less frequent dosing after the initial 24-week treatment phase. Regeneron Pharma has aggressively marketed Eylea® as “lasting longer” than Lucentis®, thus requiring fewer injections per year. Although the actual number of injections only marginally favors Eylea® at roughly 8-9 per eye per year vs. Lucentis® at 10-12 per eye per year, the advantage has been enough – with similar efficacy – to generate over $2.0 billion in sales in AMD and DME, combined.
Therefore, WE CONCLUDE that a significant market opportunity exists for a product with equal efficacy to the anti-VEGF therapies, assuming there are improvements in both systemic safety and dosing frequency.
Enter iCo Therapeutics – iCo-007
Vancouver, Canada based iCo Therapeutics is developing iCo-007, a second generation antisense drug in-licensed from ISIS Pharmaceuticals in 2005 for the treatment of diabetic macular edema. iCo-007 is designed to block c-Raf (also known as Raf-1), a protein kinase involved in transmitting the signal generated when VEGF and other growth factors believed to be important in DME bind to their cell surface receptors. This binding initiates a signaling cascade response resulting in aberrant cell growth and/or angiogenesis. The raf gene family includes three highly conserved genes termed A-raf, B-raf and c-raf. Raf genes encode protein kinases that play important regulatory roles in signal transduction processes that regulate cell proliferation and angiogenesis (US 20130028889). Research shows that aberrant cell growth and/or angiogenesis has been associated with a variety of ocular diseases and disorders, including, but not limited to, macular edema, macular degeneration, diabetic retinopathy, and retinopathy of prematurity. By targeting c-raf, the antisense oligonucleotide, iCo-007, is designed to inhibit raf gene expression and cell growth, leading to reduced ocular neovascularization.
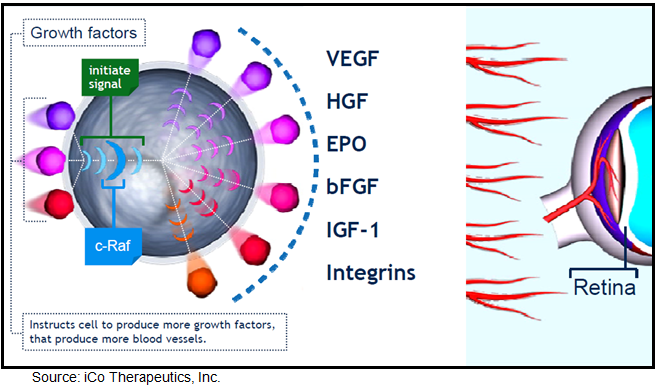{kind=link}
By blocking multiple pathways involved in the pathogenesis of DME, iCo-007 could potentially provide greater efficacy than VEGF blockers such as Lucentis® and Eylea®. Work done by Mohammad G. et al, 2011 and published in the April 2011 (Vol 15(4): 357-364) issue of Expert Opinion Therapeutic Targets qualifies the role of Raf-mediated signaling pathways as they relate to diabetic retinopathy. The authors conclude that Raf kinase is involved in a variety of functions, including the cell cycle, proliferation and apoptosis. In animal models of diabetic retinopathy, Raf kinase is activated in the retina and its microvasculature. Activated Raf kinase is associated with increased apoptosis of retinal capillary cells, the process that precedes the development of retinal histopathology, and inhibition of Raf kinase ameliorates apoptosis.
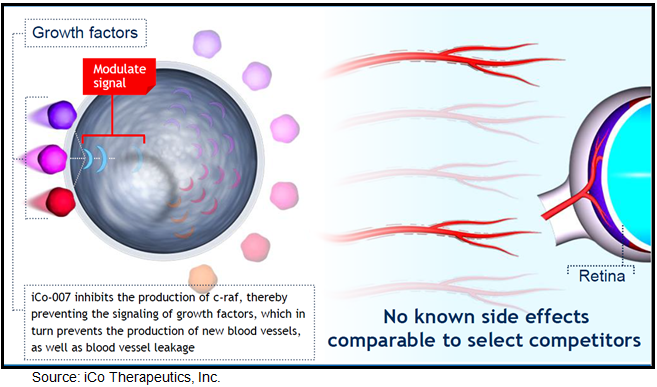{kind=link}
In July 2009, Hnik et al published a detailed analysis of the mechanism of action for iCo-007 in the Journal of Diabetes Science and Technology (Vol.3, Issue 4:924-931), titled Antisense Oligonucleotide Therapy in Diabetic Retinopathy. The authors conclude that a number of growth factors critical for neo-vascularization and leakage signal through the MAP kinase cascade, which includes c-Raf kinase, and are involved in cellular processes regulating proliferation, differentiation, and apoptosis. Other growth factors, such as erythropoietin, hepatocyte growth factor, basic fibroblast growth factor, and others, may play important roles in the etiology of diabetic retinopathy and DME.
Inhibiting a downstream target such as c-Raf kinase may prove to be a more effective treatment strategy by down regulating multiple growth factors as opposed to targeting individual growth factors only. An antisense oligonucleotide, such as iCo-007, delivered via an intravitreous injection seems to be a reasonable strategy in the treatment of retinal diseases given that experimental treatments targeting VEGF have shown clinical efficacy, and down regulating multiple growth factors that seem to play a critical role in the process of ocular angiogenesis and leakage hold scientific merit.
Hnik P. et al conclude that iCo-007 may result in a more potent anatomical effect (retinal thickness), as well as a functional effect (vision) than current anti-VEGF therapy. The authors believe that the benefits of iCo-007 include an extended half-life, resulting in less frequent intravitreal drug administration, resistance to molecule degradation, a good safety profile, and, due to a differing mechanism of action, the possibility of adjunct therapy to other agents or procedures.
Therefore, WE CONCLUDE based on a preponderance of evidence in the scientific literature (1, 2, 3, 4, 5) that the mechanism of action for iCo-007 has significant merit, and intravitreal injection of an antisense oligonucleotide is both clinically and commercially validated by the approval of Vitravene (6).
…iCo-007 Preclinical Data…
In preclinical rabbit and monkey studies, iCo-007 demonstrated an extended half-life (t½) in the retina between 6 and 8 weeks. The company has stated that it believes the results of these studies may support the potential of iCo-007 to be administered in humans at intervals of once every three to six months (source).
Results of single and multiple dosing regimens show good tissue concentration and slow clearance from the retina. We believe this could be an important differentiator in the marketing of the drug given intravitreal injection remains a source of anxiety for patients with eye disease.
…iCo-007 Phase 1 Data…
In May 2010, iCo Therapeutics announced results from an open-label Phase 1 clinical trial examining the safety and efficacy of 110 ug, 350 ug, 700 ug, or 1000 ug of iCo-007 administered via single intravitreal injection in 15 patients with diffuse macular edema. Diffuse DME is considered more difficult to treat than focal DME, which involves a smaller portion of the retina. All of these patients were non-responders to previous therapies, including photocoagulation, steroids, and anti-VEGF therapeutics such as Avastin® and Lucentis®. All patients had either Type-1 or Type-2 diabetes, with BCVA at baseline of 60-15 letters (ETDRS) or approximately 20/63 to 20/500 (Snellen). Visits took place at Day 0, Day 3, Day 7, Week 2, Week 4, Week 8, Week 12, Week 18 and Week 24. At all visits, patients were evaluated for changes in central retinal thickness (a measure of edema) and visual acuity. Among the 12 patients available for evaluation at 24 weeks (5.6 months), changes in retinal thickness in responsive patients were -149 to -743 uM, with a mean value of -169 uM. Data from the Eylea® COPERNICUS study, 2 mg every four weeks resulted in a range of -127 to -195 uM reduction in central retinal thickness after 24 weeks after six injections (source: Brown DM, et al, 2013).
Optical coherence tomography images for some of these patients show continuing improvements in macular edema between Weeks 18 and 24 of the study, supporting the potential for an extended period between injections.
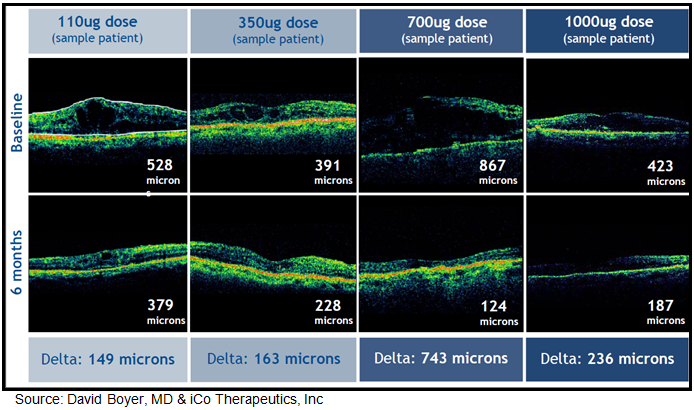{kind=link}
Thirteen patients were available for evaluation of visual acuity at the end of the 24-week treatment period; 9 of these had stable or improved vision (defined as a -5 letters BCVA or better compared to baseline) and three had a 5-letter or greater increase relative to baseline. From a safety standpoint, we note iCo-007 was not detectable in blood plasma, as expected from the low dose and its local administration into the eye. Additionally, there were no drug-related SAEs as assessed by the investigator.
We believe the efficacy results for this trial compare very well to Lucentis®, Eylea®, and Iluvien®. It is difficult to draw a firm comparison between iCo-007 and the other agents because of the different intervals between treatment and efficacy evaluation, and the lack of a disclosed mean change in visual acuity. However, the overall effect on macular thickness for iCo-007 looks impressive. That being said, the effect on visual acuity appears smaller than many of the other agents. However, we emphasize that this is a comparison of treatment effects seen six months after a single injection of iCo-007 to those seen after a 6 month, multi-injection treatment regimen of the other agents (many patients had end-stage disease).
The data is at least compatible with the hypothesis that iCo-007 can provide results comparable to those obtained with Lucentis® and Eylea®, but with a longer interval between injections. In our view, this represents an important commercial advantage. For example, Iluvien® allows very long intervals between injections, but is saddled with steroid side effects including a clinically significant increase in ocular pressure (which predisposes to glaucoma) and cataracts, and analysts that cover Alimera Sciences have estimated Iluvien® at $20 million in U.S. sales (source: Zacks Consensus).
…The Phase 2 iDEAL Study…
In March 2012, iCo Therapeutics initiated enrollment in iDEAL (NCT01565148), a Phase 2 study designed to explore whether varying combinations and concentrations of iCo-007 are effective in improving visual acuity in people with diabetic macular edema. The trial is co-sponsored by the Juvenile Diabetes Research Foundation (JDRF), which provides certain negotiated cost discounts and improved pricing metrics for iCo Therapeutics. For instance, we believe the cost of iDEAL to iCo Therapeutics is around $6 million, whereas without JDRF support the trial might have costs upward of $10 million.
To be eligible for the trial, participants must have Type-1 or Type-2 diabetes, baseline best corrected visual acuity between 20/32 and 20/320 and DME with central retinal thickness equal to or greater than 250 microns measured by optical coherence tomography (OCT). In iDEAL, patients will receive repeated intravitreal injections of iCo-007 as a monotherapy and in combination with ranibizumab (Lucentis®) or laser photocoagulation, randomized into one of four treatment groups noted below:
- Intravitreal dosing of 350 µg iCo-007 at baseline and Month-4
- Intravitreal dosing of 700 µg iCo-007 at baseline and Month-4
- Intravitreal dosing of 350 µg iCo-007 at baseline + laser photocoagulation Day-7, then another intravitreal dosing of 350 µg iCo-007 at Month-4 + laser photocoagulation at seven days later (if necessary).
- Intravitreal dosing of 0.5 mg ranibizumab (Lucentis®) at baseline + 350 µg iCo-007 at Day-14, then a repeat dose of 0.5 mg ranibizumab at Month-4 + 350 µg iCo-007 14 days later.
The primary endpoint of the iDEAL study is mean change in visual acuity (VA) from baseline to month eight. Primary safety outcome measures include the number of participants in a given study arm experiencing the same drug-related serious adverse event as a measure of safety and tolerability at month 12 and safety of repeated iCo-007 intravitreal injections in treatment of subjects with DME as monotherapy and in combination with ranibizumab or laser photocoagulation throughout the entire study. Secondary outcome measures include change in VA from baseline to month 12, change in retinal thickness measured by OCT from baseline to month eight and 12, duration of iCo-007 treatment effect during the 12-month follow-up period as measured by VA and OCT thickness, and then peak plasma concentration (CMAX) of iCo-007 after multiple injections.
The mid-point of the trial was reached in January 2013, and the company announced no serious drug-related adverse events among patients receiving repeat doses of iCo-007. In June 2013, the company announced that enrollment was completed at 187 patients (randomized). Recruitment took place at roughly 28 clinical sites across the United States. The iDEAL study is a multi-center study chaired by Quan Dong Nguyen, MD, MSc, Professor and Chair of Ophthalmology and Director of the Stanley M. Truhlsen Eye Institute at University of Nebraska Medical Center (UNMC). In addition, the Retinal Imaging Research and Reading Center (RIRRC) based at the UNMC serves as the Reading Center for the iDEAL Study.
We are expecting top-line results from the iDEAL study in April 2014. This data will include the initial analysis of the primary endpoint at month eight. The trial is scheduled to continue to month twelve, where a full analysis of the primary endpoint at month eight will be complemented by additional analysis at month twelve and a full safety set analysis of all patients receiving iCo-007, laser photocoagulation, and Lucentis®. We expect this data around the September / October time frame.
…What’s Next…
Results from the iDEAL study should help management plan the next stage in development for iCo-007. The results from the Eylea® VISTA-DME study have set the bar high. However, the potential for less frequent dosing (quarterly vs. monthly or every other month) present a very interesting market share grabbing opportunity if iCo-007 can demonstrate 10-12 lines of improvement on BCVA similar to Eylea®. However, other interesting development pathways exist for iCo Therapeutics should the results fall shy of Eylea’s impressive data from VISTA. Specifically, results in anti-VEGF non-responders will be very important for the future development of the drug. Data shows that approximately 30-40% of DME (and AMD) patients are non-responders to drugs like Lucentis® and Eylea® (7, 8).
For iCo Therapeutics, the pending results of the iDEAL study represent a major potential inflection point in the company’s valuation. If results from iDEAL are positive, iCo is sitting on a potential blockbuster drug. The trajectory for U.S. sales of Eylea® (consensus for 2014 sales is $1.8 billion) has been extremely impressive given the only modest improvement in efficacy over Lucentis® and Avastin®. Uptake is being driven by improvements in dosing and safety. The potential profile for iCo-007 offers further improvements in both over Eylea®. It comes down to whether or not the iDEAL study offers the data on the efficacy side to validate the thesis.
The next step after the full iDEAL data in September / October 2014 is to schedule a meeting with the U.S. FDA to go over the results and outline the protocol for the Phase 3 studies. As noted above, it’s a little early to predict what the potential Phase 3 studies will look like until we see the data from iDEAL. iCo Therapeutics could be looking at a broad monotherapy superiority or non-inferiority study to Lucentis®, or a combination therapy trial with anti-VEGF, or a smaller program in anti-VEGF non-responders.
…Market Opportunity For iCo-007…
According to the American Diabetes Association, 25.8 million Americans have diabetes. However, according to the National Diabetes Education Program, as many as 79 million are pre-diabetic. By 2025, the ADA expects over 40 million Americans to have diabetes. The primary risk factor for DME is dyslipidemia. Above we noted a prevalence study suggesting approximately 1.8 million people in the U.S. with DME. Data suggest the population with diabetic retinopathy is as much as 4-times the size of the DME population.
Sales of Lucentis®, primarily indicated for age-related macular degeneration, totaled roughly $4.0 billion in 2013. Analysts that cover Roche estimate approximately 2/3rds of the sales are in AMD, with the remaining 1/3rd in DME. For Regeneron’s Eylea®, sales in 2013 were $1.8 billion, of which roughly 1/3rd is in DME. We estimate the total size of the DME market at roughly $5.0 billion
As noted above, the driving factors for use of anti-VEGF therapies are: 1) superior efficacy vs. macular laser or corticosteroid injection, 2) ability to restore or improve vision loss, 3) less frequent dosing, 4) system safety and tolerability, and 5) cost. It is difficult to forecast sales of iCo-007 prior to the presentation and our analysis of the Phase 2 data from iDEAL. Following the results of iDEAL, we believe iCo Therapeutics would like to move quickly into partnership discussions on a regional basis for the pivotal registration trials and commercial sales of the product post approval.
What seems clear to us is that with similar efficacy comparable to drugs like Lucentis® and Eylea®, along with better systemic safety and the potential for less frequent dosing, iCo-007 could be a $1.0 billion drug in DME. As a combination therapy or with a label for anti-VEGF non-responders, iCo-007 is still a $500 million drug.
Intellectual Property
The composition of matter patents on iCo-007 expire in 2014-2016, which is clearly before the compound can be brought to market given its current stage of development. Other protection is provided by the ISIS patents on second-generation antisense technology, which has been licensed exclusively to iCo Therapeutics. Specifically, intellectual property rights for iCo-007 are protected in that under the licensing agreement with iCo, ISIS will not develop or commercialize or grant any sub-license or other rights to a third party to develop or commercialize any antisense RNA or analog thereof that directly inhibits c-Raf kinase (the target for iCo-007) expression or translation. These process chemistry patents are expected to run to 2023 or later, but this is still only 5 years after the expected approval date of iCo-007, and these patents would presumably not be eligible for patent term extension under the Hatch-Waxman amendment.
The company reports that it has applied for additional patents to protect iCo-007. One such patent has been published on the USPTO website (the usual delay after filing being 18 months). Application 13/577,219 claims a method of treating macular edema, comprising administering an anti-c-Raf oligonucleotide no more frequently than once every 90 days. Given the novelty and utility of such infrequent administration, we believe there is at least a 50% chance that this application could be granted and successfully defended in court. We expect, however, that it will be a few years until a final decision is available from the USPTO, as the application was only just filed in 2011. If granted and successfully defended, protection would be available for 15 years or more post-approval.
For purposes of our valuation, we assume that the exclusivity on iCo-007 will be limited to the data exclusivity provided by the Hatch-Waxman Act. The terms of the Act forbid the filing of an NDA on a new chemical entity for 5 years post-approval. These 5 years can be extended by 6 months for performing pediatric studies. Adding to this the approximately 2 years required by the FDA to process a typical ANDA application, we estimate the marketing exclusivity available to iCo on iCo-007 will be around 7 to 8 years post approval.
iCo Therapeutics Very Attractive
iCo Therapeutics shares currently trade with a market capitalization of only $30 million for the U.S. OTCQX listed shares. iCo Therapeutics also trades on the TSX-Venture in Canada. There are four key points that lead us to delve deeper into the iCo story.
- Firstly, in November 2013, the company saw the final exercising of roughly 3.207 million warrants at $0.30 dating back to the November 2011 financing, raising gross proceeds of approximately $0.962 million. We were pleased to see the company pull in this extra cash with existing investors.
- Secondly, in December 2013, the company’s shares began trading on the OTCQX, the highest tier of the OTC Market, under the ticker symbol: ICOTF. We believe the QX listing will help iCo Therapeutics gain greater exposure and increased liquidity with biotech investors in the U.S.
- Thirdly, in late January 2014, the company raised gross proceeds of CAD-$6.75 million through the issuance of 16.206 million shares of common stock at $0.4165 per share. We modeled that iCo Therapeutics exited 2013 with roughly $1.5 million in cash. Adding into this new net $6.3 million cash from the equity financing should put cash and investments at roughly $6.0 million as of March 31, 2014. This effectively eliminates any new financing risk prior to the release of the iDEAL data and creates a clear catalyst to our fourth key point. We note that previous investors in the company, the Special Situations Fund III QP, L.P. and Special Situations Life Sciences Fund, L.P., increased their position in this offering and now own 16.5% of the outstanding shares.
- Fourthly, top-line results from the iDEAL study are expected in April 2014, creating a clear valuation inflection point for investors if the data are positive.
…Value Heavily Dependent On iDEAL…
Our analysis of iCo-007 began with a thorough analysis of antisense drug development, and that acknowledgement of antisense technology has not lived up to the hype that ensued over two decades ago when the technology was first brought public. Nevertheless, through trial and error, it now seems abundantly clear that success of an antisense drug is dependent on delivery and target location. For iCo, delivery of an antisense drug through intravitreal injection has been fully validated from a clinical and commercial standpoint by the approval and launch of Vitravene at Novartis. Treatment of DME represents a large market opportunity, and the mechanism of action of the current leading therapies, anti-VEGF, has been proven highly successful by intravitreal injections of drugs like Avastin®, Lucentis®, and Eylea®. More specifically, however, the mechanism of action of iCo-007, upstream blockage of the protein kinase, c-Raf, a signal transmitter for VEGF and other growth factors, hold significant scientific merit (1, 2, 3, 4, 5). Our analysis of published literature leads us to conclude that the Raf genes play an important regulatory role in signal transduction processes that regulate cell proliferation and angiogenesis. Patents have been awarded protecting such technology, and preclinical and Phase 1 data yield highly encouraging results for iCo-007.
For the purpose of establishing valuation of iCo Therapeutics, we estimate 50% likelihood that the iDEAL trial will demonstrate acceptable safety and an efficacy profile comparable to Lucentis® after eight months (two doses). The results from the Eylea® VISTA-DME study have set the bar high. However, the potential for less frequent dosing (quarterly vs. monthly or every other month) present a very interesting market share grabbing opportunity if iCo-007 can demonstrate 10-12 lines of improvement on BCVA similar to Eylea®. However, other interesting development pathways exist for iCo Therapeutics should the results fall shy of Eylea’s impressive data from VISTA. Specifically, results in anti-VEGF non-responders will be very important for the future development of the drug. Research shows that approximately 30-40% of DME (and AMD) patients are non-responders to drugs like Lucentis® and Eylea®.
For iCo Therapeutics, the pending results of the iDEAL study represent a major potential inflection point in the company’s valuation. If results from iDEAL are positive, iCo is sitting on a potential $1+ billion drug. A label for anti-VEGF non-responders still offers a potential $500 million opportunity. Assuming a path forward, we believe that the company will likely seek a development partner for the U.S. Phase 3 registration program. Such a deal may be signed with $50 million in upfront licensing payments to iCo Therapeutics, with a potential backend regulatory and sales milestone payment totaling $500 million. We also project any such deal would pay iCo a tiered royalty on sales, ranging from the low-teens to low-twenty percent from the commercialization partner.
Assuming U.S. approval in 2018, with the aforementioned peak sales of $1 billion and the out-licensing terms outlined above, with 50% likelihood of a positive iDEAL trial and a 25% discount rate, we see the iCo-007 NPV at $80 million. Gaining of additional patent applications that extend exclusivity of the molecule beyond 2023 could add as much as $50 million in value to our NPV calculation. We remind investors that our NPV calculation factors in terms of the original license agreement with ISIS back in 2005, and includes up to $22 million in developmental milestones and unspecified royalties on sales (which we model at 6%).
…iCo-008 iCo-009 Represent Meaningful Upside…
Beyond iCo-007 and the pending outcome of the iDEAL study, iCo Therapeutics has two other potential revenue drivers that warrant inclusion in our sum-of-parts valuation model. The first is iCo-008, a first-in-class fully human IgG4 monoclonal antibody discovered using Cambridge Antibody Technology’s phage display technology, and licensed to iCo in January 2007. The mechanism of action of iCo-008, also known as bertilimumab, has been described in detail by numerous authors in peer-reviewed papers we reviewed for preparation of our report.
Preclinical work show high affinity and specificity for eotaxin-1, a pro-inflammatory cytokine that plays a critical role in mast cell degranulation and the development of an inflammatory condition known as eosinophilia. Clinical and preclinical evidence proves that by blocking eotaxin-1 can result in effective inhibition of early phase mast cell activation as well as late phase eosinophilia. iCo Therapeutics believes this mechanism of action has clinical utility for the treatment of vernal keratoconjunctivitis (VKC) and wet age-related macular degeneration (AMD).
Beyond ocular indications, iCo Therapeutics has out-licensed bertilimumab to Israeli-based Immune Pharmaceuticals (OTCQX:IMNP) for the treatment of Irritable Bowel Disease (IBD) and Bullous Pemphigoid (BP). The clinical data in all indication is a little early-stage, and thus forecasting potential peak sales and probabilities of approval is difficult. However based on modest success in VKC and/or AMD, and similar blockbuster success by Immune Pharma in IBD or BP, we think iCo-008 is worth $25 million.
iCo Therapeutics also owns the worldwide exclusive rights to an oral Amphotericin B delivery system for life-threatening infections. Amphotericin B (AmpB), the gold standard for systemic antifungal drugs, is one example of a well-established, highly efficacious systemic antifungal drug that has a 50-year history of intravenous therapy. iCo’s candidate is the company’s oral lipid capsule formulation of AmpB. The company acquired the exclusive worldwide rights to the drug back in May 2008 from the Wasan Labs at the University of British Columbia. In November 2013, U.S. patent #8,592,382 was issued for the Oral Amphotericin B platform providing protection around oral delivery of the drug.
iCo Therapeutics has progressed into in vitro testing with study partners in Montreal, and will examine the role of this formulation in targeting latent HIV reservoirs, which remain in individuals despite enormous therapeutic advances in the treatment of HIV/AIDS. Recruitment of eight HIV-infected subjects successfully treated with highly active antiretroviral therapy (HAART) with detectable latent viral reservoir is expected to be complete in the first half of 2014. In September 2010, iCo Therapeutics was granted Orphan Drug status by the U.S. FDA for the treatment of Visceral Leishmaniasis (VL).
Orphan designation qualifies iCo for tax and marketing incentives, which can include tax credits for clinical research, study design support, exemption from application-filing fees, grant funding for clinical trials, and seven years of marketing exclusivity after the approval of the drug.
As noted above, animal model studies from 2008 showed a 99% knock-down effect of parasitic infection caused by VL with 10 and 20 mg/kg of iCo-009 oral administration BID for one week. We have assumed very modest success for iCo-009 in our valuation model, concluding that without more substantial human clinical data, the candidate is worth roughly $10 million in value.
iCo Therapeutics shares are currently trading with a market value of only $45 million on a fully-diluted basis (based on $0.40 per share). Chief competitors in this space, Ohr Pharmaceuticals, Alimera Sciences, and pSivida Corp. are all trading at significant premiums to iCo.
Sum-of-parts analysis tells us that iCo’s stock has 125% upside based on a 50% probability of success of iDEAL in April 2014. Comparator analysis tell us that the shares could be worth as much as $3.00, or +633% upside, if iDEAL hits on the full data analysis in September / October 2014. As such, we see meaningful upside in owning the stock at this level.
Risks To Consider
Despite what we see as a meaningfully undervalued stock, there are certain risks to consider prior to an investment in iCo Therapeutics shares.
- iCo-007 Fails: The primary risk for investors in buying iCo Therapeutics’ stock prior to the release of the top-line data from the iDEAL study in April 2014 is that the drug fails to demonstrate meaningful efficacy on BCVA when compared to Lucentis® or Eylea®. iCo-007 represents $80 million in our sum-of-parts valuation analysis. Failure of the drug would force us to slash our price target to $35 million on a fully-diluted basis. This equates to $0.30 per share. We suspect the shares may trade below this level given that in our conversations with investors, limited value at all is being assigned to iCo-008 or iCo-009.
- New competition: Ohr Pharmaceuticals (OHRP) is developing Squalamine, an anti-angiogenic twice weekly eye drop currently in a Phase 2 study for the treatment of AMD, with future plans to expand into DME if it works. Squalamine’s mechanism of action is similar to Lucentis® and Eylea® in its targeting of VEGF, PDGF, and other growth factors that lead to angiogenesis. The clear benefit of self-administered eye drops vs. once weekly, bi-weekly, or even monthly intravitreal injections by an ophthalmologist cannot be overstated. The data on Squalamine remains early-stage. However, if successful and eventually commercialized, the drug could dominate the market with similar efficacy to the anti-VEGF drugs like Lucentis® and Eylea®. Ohr Pharmaceuticals has an impressive Ophthalmic Advisory Board, which includes David Boyer, MD who published on iCo’s preclinical and Phase 1 data. We suspect that if Squalamine shows promise in the ongoing Phase 2 study, designed as a rescue therapy assessment for new AMD patients on Lucentis®, Squalamine could end up in the hands of a major player in the ophthalmology field.
- New Financings: We model iCo Therapeutics exiting the calendar first quarter 2014 with roughly $6.0 million in cash. The company’s stock in now publicly traded, Immune Pharma is worth another $2+ million. With an operating burn of around $1.5 million per quarter, we believe iCo will require new cash to fund operations later in 2014. If the top-line results from iDEAL are positive, we suspect that iCo will be able to secure new funding at meaningfully higher levels. However, we do not believe the company will be able to secure a development partnership for iCo-007 until after the full results from iDEAL have been presented, which is expected to take place in September or October 2014. This is around the time we suspect the company will require new cash if they do not act on the top-line results from iDEAL this spring or summer.
Conclusion Recommendation
Sum-of-parts modeling calculates that iCo Therapeutics’ shares are meaningfully undervalued at today’s market value of only $30 million on a basic share count basis. We find this bafflingly low. We note this value does not include some 28.6 million warrants at or very near in-the-money. This could add another $15 million market value, but also $11 million in cash to the company if exercised.
Our analysis finds that the shares should be valued more in the $100-105 million range, or around $0.90 per share on a fully-diluted basis based on a 50% probability of success of the iDEAL trial. This represents tremendous upside to investors at today’s price of only $0.39 per share. However, investors need to be aware that approximately 80% of our valuation is coming from iCo-007 for the treatment of DME (and off-label use in AMD). Pipeline products iCo-008 and iCo-009 represent upside to our valuation, and some downside protection should iDEAL fail. We expect top-line results from the iDEAL study are expected in April 2014.
It is clear there is dramatically more upside in the shares based on the results of iDEAL than downside. We suspect that the failure of iDEAL hits the shares by 50%, whereas success could re-value them higher at 600% in time. This type of favorable risk / reward should be attractive to most investors.
We believe there will be a substantial multi-month run in the shares to the full data in September / October 2014 if the top-line results in April 2014 look solid. So we would advise keeping an eye on the shares for the top-line results in April 2014. We suspect significant investor attention to come to iCo in the next two months prior to the data. A prudent strategy might be to establish a position today, and then fill the rest of the order if the data from iDEAL in April 2014 impresses.
Disclosure: I have no positions in any stocks mentioned, and no plans to initiate any positions within the next 72 hours. I wrote this article myself, and it expresses my own opinions. I am not receiving compensation for it (other than from Seeking Alpha). I have no business relationship with any company whose stock is mentioned in this article.
ICO THERAPEUTICS (ICOTF) news: Ico Therapeutics: Near-Term Catalyst ...
Không có nhận xét nào:
Đăng nhận xét